Study sheds light on lung cancer therapy
Lung adenocarcinoma has been the leading cause of cancer death in Hong Kong. Though the causes of lung adenocarcinoma are complex, many studies have revealed that alterations to transcriptomes exert significant effects on its progression. A research team led by Professor Chan Ting-fung of the School of Life Sciences has developed a novel computational method, LAFITE (Low-abundance Aware Full-length Isoform clusTEr), and identified thousands of low-abundance full-length RNA transcripts in lung adenocarcinoma cell lines that were unknown using existing technologies.

A new, rare transcript isoform was also found in a cancer-driving gene that significantly correlates with patient survival and tumour cell migration in lung adenocarcinoma, furthering studies of its progression. The findings have recently been published in the prestigious journal Advanced Science.
Different tissues in an organism share the same genome, but they differ markedly in the transcriptome–the composition of expressed RNA transcripts. Previous studies revealed that the majority of expressed transcripts are present at low levels (so-called “low-abundance”) while they act as important regulators in various biological processes, including metabolic processes and cancer progression.
RNA-sequencing (RNA-seq) has now been widely used in many biological and clinical studies, but performs unsatisfactorily in identifying low expression or full-length transcripts. It fragments and copies RNA molecules to increase the quantity, then reconstructs the transcripts for easier identification of high-copy, active short-length transcripts, but low-copy full-length transcripts that exhibit low expression are difficult to identify.
Higher eukaryotes including humans can generate multiple transcript isoforms from the same gene by RNA splicing. However, RNA-seq cannot unambiguously identify individual transcript isoforms. As a result, the present research on transcriptomes has mainly focused on the characterisation of gene levels but not transcript levels, leading to ignorance of many transcripts at low expression.
Although the current third-generation sequencing technique Oxford Nanopore Technologies can capture native RNA transcripts and perform better at achieving low-abundance, full-length transcript isoform identification, the Nanopore DRS data to which this approach was applied is notoriously noisy and contains many intrinsic errors that reduce its accuracy.
Supported by the Hong Kong Research Grants Council Area of Excellence Scheme and Collaborative Research Fund, Professor Chan’s team has developed LAFITE, a novel computational method that is tailored for processing Nanopore DRS data to detect full-length isoforms. It outperformed all existing computational methods with a higher sensitivity in low-abundance RNA transcripts.
The team applied LAFITE to study the Nanopore DRS data from four lung adenocarcinoma cell lines, successfully identified a novel low-abundance RNA transcript isoform from the cancer gene AKT1 and demonstrated it to be functional in lung cancer cell lines in a way that significantly correlates with patient survival, promoting tumour cell migration in lung adenocarcinoma.
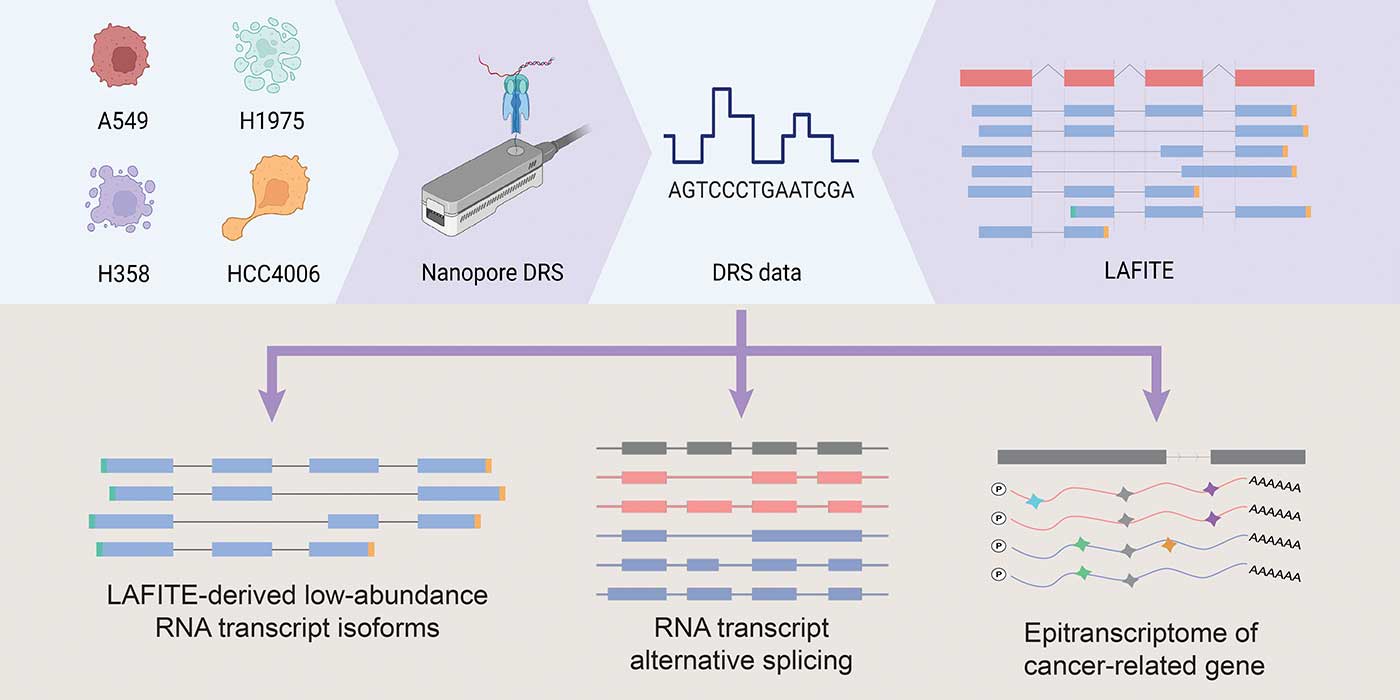
Using LAFITE, the team also discovered thousands of low-abundance transcripts overlooked by previous studies, providing a full-length transcriptome of lung adenocarcinoma. It will be a valuable resource for scientists to interpret the underlying mechanism related to the formation, migration and progression of lung cancer. The team anticipates that LAFITE will be applicable to other cancers such as colorectal cancer, and to further studies on drug resistance of cancer cells for drug advancement.
Professor Chan added, “A comprehensive characterisation of transcript isoforms of individual genes may renew our understanding of their biological functions. LAFITE provides researchers with a new solution to reassess gene function by identifying all expressed transcripts in a truly comprehensive manner, and highlights the importance of transcript-level analysis in a transcriptomic study.”
The full paper can be read here.